
前言
“你把表达量和差异表达的表格给我就行了”
花那么多钱就换两张表格,你的良心不会痛么!(当年表达芯片都这么做的也没见谁良心痛啊)
当然,毕竟别人不一定靠这个吃饭,但 (找不到工作的) 学生物的你,稍微了解一下测序的分析流程还是值得的,毕竟技多不压身嘛。
所以这一部分主要介绍转录组测序的分析流程和原理,从拿到原始数据开始,讲到KEGG/Gene Ontology等功能注释,顺便推荐一下常用软件。字数所限,这一篇先讲 (不生成文章能用的图表的) Data Cleaning和比对,如果只想知道怎么看懂文献里面的结果,可以直接等下一篇了。
流程概览
转录组测序的分析流程大致可以分成三类,包括基因组比对(Genome mapping)、转录组比对(Transcriptome mapping)、转录组组装(Reference-free assembly),见下图。其中第三种主要是用于分析没有参考基因组和基因注释的物种,应用场合较少且不适合新手入门。对于人、小鼠、大鼠等模式物种,通常用前两种方法进行分析。虽然转录组比对相关软件和流程同样层出不穷,但对于基因组信息较为完善的模式物种,推荐使用基因组比对的方式进行分析,具体原因下文的“比对”部分会有说明。我们下面也主要对基因组比对的方法进行介绍。
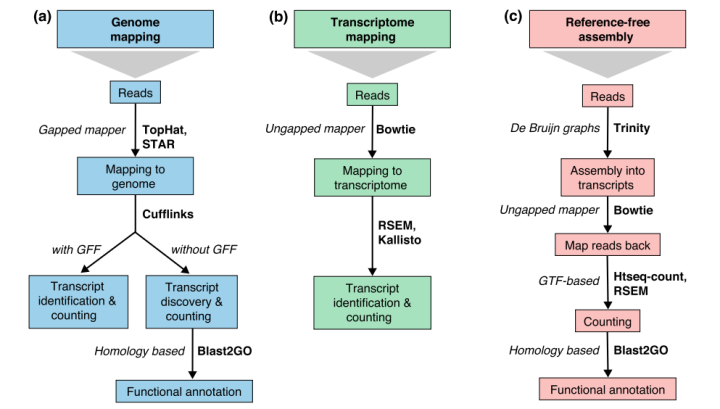
图片来源:https://www.ncbi.nlm.nih.gov/pubmed/26813401
1. Data Cleaning
从原始数据(Raw Data)到干净数据(Clean Data)的过程,有人翻译成“数据清洗”,实在叫不习惯,那我就不翻译了。
Illumina测序仪下机的数据通常为Bcl格式,是将同一个测序通道(Lane)所有样品的数据混杂在一起的,所以公司一般不会提供Bcl文件。测序公司使用Illumina官方出品的Bcl2FastQ软件,根据Index序列分割转换成每个样品的FastQ文件,打开长这样:

每一条序列(read)包含四行,第一行是read的ID,第二行是序列,第四行是序列中每个碱基的测序质量(更具体的细节可参考FASTQ format - Wikipedia)。
原始数据没法直接分析,是因为部分reads测序质量较低,可能会误导后续结果,因此需要对低质量碱基太多或N(未能识别的碱基)太多的reads进行去除;此外,部分测序文库的插入片段太短,导致测到两侧的接头序列(请参考上一篇的测序文库结构图理解),这些序列接头也需要从reads中去除。最后,我们也会对清洗前后的Raw Data和Clean Data进行评估,评估内容包括碱基质量、序列长度、碱基比例、GC含量、重复序列、Kmers等(详情请参考FastQC说明文档FastQC A Quality Control tool for High Throughput Sequence Data)。
最后说一句,其实大多数测序公司都会提供Clean Data的。
#常用软件#
我以前都是用cutadapt + FASTX-Toolkit的组合,直到同事们给我推荐了Trim Galore,质量评估使用FastQC。
2. 比对
由于二代测序的reads长度通常介于50~300个碱基,因此即便使用双端测序,也基本不可能覆盖完整的mRNA转录本,因此想直接用FastQ文件从头分析测到了哪些转录本需要非常复杂的分析和计算。好在通常情况下,公共数据库已经提供了测序样品的基因组和转录本的序列。因此我们只需要知道,每一条reads来自哪一条转录本就可以了,这个将reads与参考(Reference)基因组/转录组的序列进行比较和匹配的过程,我们通常称之为“比对”(文献中提到的read alignment和mapping通常说的都是这个)。
正如前文所述,转录组测序的比对通常分为基因组比对和转录组比对两种,顾名思义,基因组比对就是把reads比对到完整的基因组序列上,而转录组比对则是把reads比对到所有已知的转录本序列上。如果不是很急或者只想知道已知转录本表达量,个人建议使用基因组比对的方法进行分析,理由如下:
① 转录组比对需要准确的已知转录本的序列,对于来自未知转录本(比如一些未被数据库收录的lncRNA)或序列不准确的reads无法正确比对;
② 与上一条类似,转录组比对不能对转录本的可变剪接进行分析,数据库中未收录的剪接位点会被直接丢弃;
③ 由于同一个基因存在不同的转录本,因此很多reads可以同时完美比对到多个转录本,reads的比对评分会偏低,可能被后续计算表达量的软件舍弃,影响后续分析(有部分软件解决了这个问题);
④ 由于与DNA测序使用的参考序列不同,因此不利于RNA和DNA数据的整合分析。
而上面的问题使用基因组比对都可以解决。
此外,值得注意的是,RNA测序并不能直接使用DNA测序常用的BWA、Bowtie等比对软件,这是由于真核生物内含子的存在,导致测到的reads并不与基因组序列完全一致(如下图所示),因此需要使用Tophat/HISAT/STAR等专门为RNA测序设计的软件进行比对。
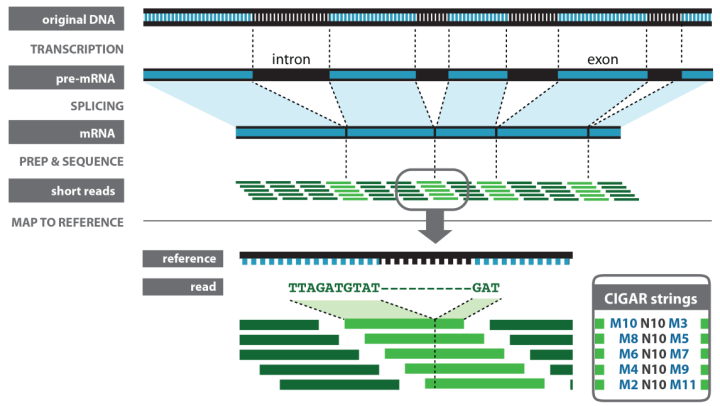
| 图片来源:[GATK | Best Practices](https://link.zhihu.com/?target=https%3A//software.broadinstitute.org/gatk/best-practices/rnaseq.php) |
比对结果会展示为BAM/SAM文件,其中BAM格式是SAM格式的二进制版本(请理解为压缩后的版本,用Samtools可以打开),打开之后长这样:
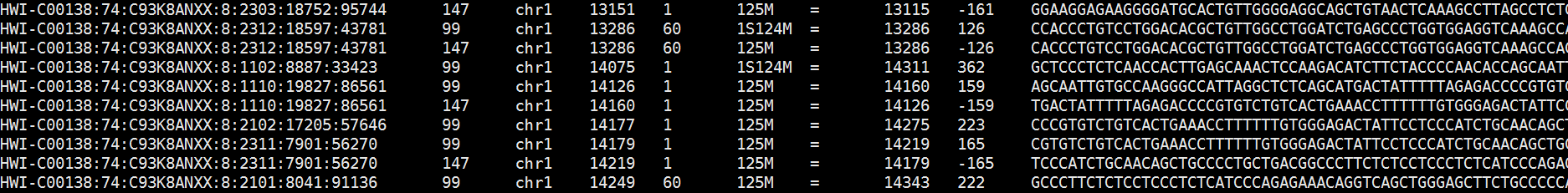
BAM文件中每行代表一条reads的比对信息,其中第一列是read的ID,第二列为FLAG(包括是否双端比对,比对位点是否唯一等信息),第三列为比对的染色体,第四列为比对的起始位置,第六列为CIGAR值,代表比对的具体方式(例60M2D80M代表60个碱基完美匹配+2个碱基缺失+80个碱基完美匹配)等等,BAM文件的具体内容可参考SAM - Genome Analysis Wiki和http://samtools.github.io/hts-specs/SAMv1.pdf(后面这个要翻墙)。
#常用软件#
基因组比对:
Tophat2:可以说是最被公认的RNA测序比对软件(实际上是在DNA比对软件Bowtie的基础上做了一个壳),相信很多做RNA测序的同学都是看着Tophat发表在Nature Protocol上的步骤一步步入门RNA测序的;
HISAT2:Tophat2的非正式升级版本(因为据说还会有Tophat3),在Tophat的算法基础了上做了大量的改进,而且克服了Tophat最大的缺点——速度慢,Nature Protocol上同样发表了操作流程;
STAR:ENCODE计划御用比对软件,权威程度可以与Tophat平起平坐,并且比对速度极快;
MapSplice:TCGA使用的比对软件,我自己没用过;
RSEM:RSEM更像一个软件包而不是一个比对软件,能够提供从比对到计算差异表达的所有步骤,由于不需要自己写代码串联不同软件生成的数据格式,因此用起来比较省时省力,值得注意的是,TCGA使用MapSplice比对后再用RSEM计算表达量,并没有直接只用RSEM原装的Bowtie的比对结果。
转录组比对:这类型的软件我用的不多,最近尝试过Nature Methods上面发表的Salmon,能从Clean reads直接算到表达量,优点是,快,非常快。然而这个软件连BAM文件都没生成,虽然只是定量的话BAM文件的确没什么用就是了…
1.5 #可选步骤# 核糖体RNA(rRNA)去除
嗯,写完2再写1.5是我不对。
如果对上一篇还有印象的话,我们曾提到,转录组测序有一种 偏贵的 使用核糖体RNA去除技术构建文库的测序。但是经常做实验的你一定知道,这种去除是没法做到100%去除rRNA的,更糟糕的是,同一批测序的每个样品,rRNA的去除效率也会有一定差别的!
由于rRNA都是非编码RNA序列,因此如果我们后续分析需要使用转录本组装的方法鉴别新的lncRNA(long non-coding RNA,长非编码RNA),这些rRNA的序列特征很容易对lncRNA的鉴定造成干扰,因此我们必须对这些rRNA序列进行去除。
当然,如果不涉及组装新lncRNA的话,rRNA的存在对分析结果的影响并不大。但如果样品间rRNA残留率差别较大,对定量的准确性会有较大影响,因此有能力的话还是建议去除rRNA序列。就算不去除,用比对软件算算rRNA序列占总数据量的比例也是好的,一旦不小心发现12G的数据里面6G都来自于rRNA…(嗯我不是教你们跟公司撕X…)
#常用软件#
核糖体去除实际上也是通过比对来进行,我在Rfam上下载rRNA的序列后,直接使用Bowtie2进行比对。
至于比对核糖体之后怎么拿到没有rRNA的FastQ文件,我不太清楚别人是怎么做的,我是用Python把没比对上的Reads的ID提取出来存成一个表格,再用Seqtk提取FastQ文件。
#########################未完待续,我们下回分解#########################
最后关于常用软件:
首先推荐OMIC TOOLS网站(https://omictools.com/rna-seq-category),上面收集了大量高通量测序相关的软件以及软件之间的对比评测文献。
其次,我写的常用软件基本基于我自己和身边同事的使用情况,以及在文献中看到的情况,并不涉及对软件自身性能的评判。比如华大基因开发的比对用软件SOAPsplice,没有提到不是说它不好,而是我的确没用过,而且用的人也比较少。
至于如何判断哪个软件更好,可以参考软件评测的文献,但没必要以此为绝对标准,例如我见到有评测说STAR完爆Tophat的,也有说Tophat完爆STAR的,而且这两篇评测都发在Nature Method上。我个人认为能一直存活下来也被广泛使用的软件大多各有千秋,对于一个没精力看源代码(也看不懂java和C++)的使用者来说,想要评估一个软件的好坏,直接参考高分杂志的使用情况来推算,可能是更靠谱的选择。
另外,长期欢迎推荐各类软件。