
GSEA定义
Gene Set Enrichment Analysis (基因集富集分析)用来评估一个预先定义的基因集的基因在与表型相关度排序的基因表中的分布趋势,从而判断其对表型的贡献。其输入数据包含两部分,一是已知功能的基因集 (可以是GO注释、MsigDB的注释或其它符合格式的基因集定义),一是表达矩阵(不能用差异基因来跑GSEA),软件会对基因根据其于表型的关联度(可以理解为表达值的变化)从大到小排序,然后判断基因集内每条注释下的基因是否富集于表型相关度排序后基因表的上部或下部,从而判断此基因集内基因的协同变化对表型变化的影响。
这与GO富集分析不同。GO富集分析是先筛选差异基因,再判断差异基因在哪些注释的通路存在富集;这涉及到阈值的设定,存在一定主观性并且只能用于表达变化较大的基因,即我们定义的显著差异基因。而GSEA则不局限于差异基因,从基因集的富集角度出发,理论上更容易囊括细微但协调性的变化对生物通路的影响。
注意:表达矩阵也可以替换成logFC的基因list,不过需要对logFC从高到低进行排序。
GSEA原理
给定一个排序的基因表L和一个预先定义的基因集S (比如编码某个代谢通路的产物的基因, 基因组上物理位置相近的基因,或同一GO注释下的基因),GSEA的目的是判断S里面的成员s在L里面是随机分布还是主要富集在L的顶部或底部。这些基因排序的依据是其在不同表型状态下的表达差异,若研究的基因集S的成员显著聚集在L的顶部或底部,则说明此基因集成员对表型的差异有贡献,也是我们关注的基因集。
GS为基因集的名字,SIZE代表该基因集下的基因总数,ES代表Enrichment score, NES代表归一化后的Enrichment score, NOM p-val代表pvalue,表征富集结果的可信度,FDR q-val代表qvalue, 是多重假设检验矫正后的p值,注意GSEA采用pvalue < 5%, qvalue < 25% 对结果进行过滤。Rank at max表示ES分数达到最大值时最后一个基因所处的位置。说的通俗一点,比如说在我们此次GSEA的5000个基因里,有3个基因对于对于cell cycle通路的ES分数有正贡献,三个基因分别位于第1,2,3位(本文开头说过,GSEA会根据基因表达差异大小对基因进行排序打分),他们对于ES分数的贡献分别为0.1,0.01,0.001,ES分数在第3位达到最大值0.111, 此时的Rank at max就为3,第三位往后的基因对cell cycle通路的ES分数是负贡献。
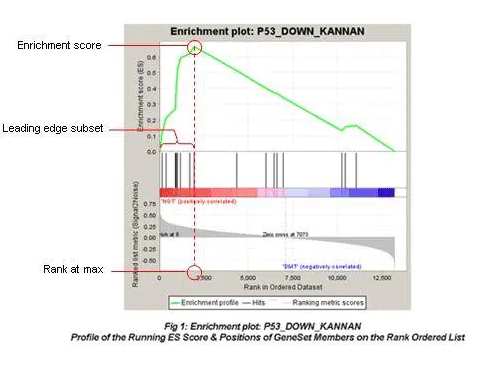
GSEA计算中几个关键概念:
- 计算富集得分 (ES, enrichment score). ES反应基因集成员
s在排序列表L的两端富集的程度。计算方式是,从基因集L的第一个基因开始,计算一个累计统计值。当遇到一个落在s里面的基因,则增加统计值。遇到一个不在s里面的基因,则降低统计值。每一步统计值增加或减少的幅度与基因的表达变化程度(更严格的是与基因和表型的关联度)是相关的。富集得分ES最后定义为最大的峰值。正值ES表示基因集在列表的顶部富集,负值ES表示基因集在列表的底部富集。 - 评估富集得分(ES)的显著性。通过基于表型而不改变基因之间关系的排列检验 (permutation test)计算观察到的富集得分(ES)出现的可能性。若样品量少,也可基于基因集做排列检验 (permutation test),计算p-value。
- 多重假设检验矫正。首先对每个基因子集
s计算得到的ES根据基因集的大小进行标准化得到Normalized Enrichment Score (NES)。随后针对NES计算假阳性率。(计算NES也有另外一种方法,是计算出的ES除以排列检验得到的所有ES的平均值) - Leading-edge subset,这些gene的排序靠前,对富集得分贡献最大的基因成员。
- 峰值的顶点为最大富集分数,
- 黑色竖条表示
s中基因在L中的位置
应用
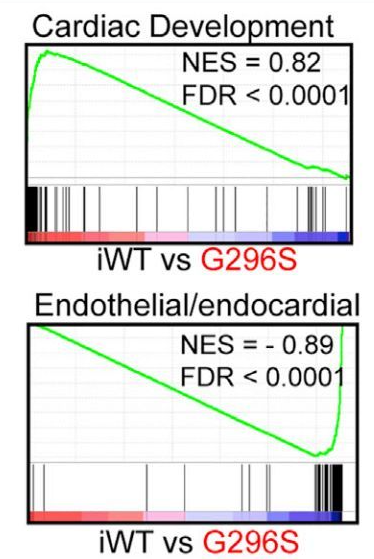
GSEA analyses of genesets for cardiac (top) and endothelial/endocardial (bottom) development. NES, normalized enrichment score. FDR, false discovery rate. Positive and negative NES indicate higher and lower expression in iwt, respectively.
文章使用的参数:
permutation = geneset, metric = Diff_of_classes, metric = weighted, #permutation = 2500
文章通过GSEA分析,发现:
与心脏发育有关的基因集 (影响心脏的收缩力、钙离子调控和新陈代谢活力等)在
iwt组 (GATA基因野生型)中普遍表达更高,而在G296S组 (GATA基因的一种突变体)中表达更低; 而对于参与内皮或内膜发育的基因集,在iwt组中表达更低,在G296S组中表达更高。 作者根据这个图和其它证据推测iwt组的心脏发育更加完善,而G296S组更倾向于心脏内皮或内膜的发育,即GATA基因的这种突变可能导致心脏内皮或内膜的过度发育而导致心脏相关疾病的产生。
在上述Cell文章中,作者更加关心参与心脏发育的基因集 (即a priori defined set of genes)与两个状态(突变体和野生型,状态的度量方式是基因表达)的关系,因此利用GSEA对其进行分析后发现,参与心脏发育 (收缩力、钙调控和新陈代谢)的基因集的表达模式更接近于iwt组的表型,而不是G296S组; 而参与心脏内皮或内膜发育的这些基因的表达模式更接近于G296S组的表型而不是iwt组的表型。
这就是GSEA分析所适用的主要场景之一。它能帮助生物学家在两种不同的生物学状态 (biological states)中,判断某一组有特定意义的基因集合的表达模式更接近于其中哪一种。因此GSEA是一种非常常见且实用的分析方法,可以将数个基因组成的基因集与整个转录组、修饰组等做出简单而清晰的关联分析。
除了对特定gene set的分析,反过来GSEA也可以用于发现两组样本从表达或其它度量水平分别与哪些特定生物学意义的基因集有显著关联,或者发现哪些基因集的表达模式或其他模式更接近于表型A、哪些更接近于表型B。这些特定的基因集合可以从GO、KEGG、Reactome、hallmark或MSigDB等基因集中获取,其中MSigDB数据库整合了上述所有基因集。研究者也可自定义gene set (即新发现的基因集或其它感兴趣的基因的集合)。
GSEA分析似乎与GO分析类似但又有所不同。GO分析更加依赖差异基因,实则是对一部分基因的分析 (忽略差异不显著的基因),而GSEA是从全体基因的表达矩阵中找出具有协同差异 (concordant differences)的基因集,故能兼顾差异较小的基因。因此二者的应用场景略有区别。另外GO富集是定性的分析,GSEA考虑到了表达或其它度量水平的值的影响。另外,对于时间序列数据或样品有定量属性时,GSEA的优势会更明显,不需要每个分组分别进行富集,直接对整体进行处理。可以类比于之前的WGCNA分析。
MSigDB数据库
-
注释数据库
category description 描述 H hallmark gene sets (效应)特征基因集合 C1 positional gene sets 位置基因集合,根据染色体位置 C2 curated gene sets (专家)共识基因集合,基于通路、文献等(包括KEGG) C3 motif gene sets 模式基因集合,主要包括microRNA和转录因子靶基因两部分 C4 computational gene sets 计算基因集合,通过挖掘癌症相关芯片数据定义的基因集合 C5 GO gene sets 基因本体论(包括BP/CC/MF) C6 oncogenic signatures 癌症特征基因集合,大部分来源于NCBI GEO 未发表芯片数据 C7 immunologic signatures 免疫相关基因集合 使用方法:
## devtools::install_github("ToledoEM/msigdf") library(msigdf) -
MsigDB目录简介:
-
H: hallmark gene sets 该类别包含了由多个已知的基因集构成的超基因集,每个H类别的基因集都对应多个基础的其他类别的基因集。比如HALLMARK_ADIPOGENESIS对应36个基因集。
-
C1: positional gene sets 该类别包含人类每条染色体上的不同cytoband区域对应的基因集合。根据不同染色体编号进行二级分类。
-
C2:curated gene sets 该类别包含了已知数据库,文献和专家支持的基因集信息,包含下面5个二级分类

以
KEGG为例,包含了186个基因集,每个基因集本质上都对应pathway 数据库里的一条通路。-
C3 : motif gene sets
该类别包含了miRNA靶基因和转录因子结合区域等基因集合,示意如下
无论是转录因子还是miRNA, 都是通过特定的motif序列来识别可以结合的区域,这些基因集合,本质上为具有相同motif序列的基因集,比如AAACCAC_MIR140这个基因集, 具有相同的AAACCACmotif, 而hsa-miR-140可以识别该motif然后进行结合,所以AAACCAC_MIR140是hsa-miR-140靶标基因的集合。
-
C4 : computational gene sets
该类别包含计算机软件预测出来的基因集合,主要是和癌症相关的基因,示意如下
-
C5 : GO gene sets
该类别包含了Gene Ontology对应的基因集合,分为以下3大类别

每个基因集对应一个GO term
-
C6 : oncogenic signatures
该类别包含已知条件处理后基因表达量发生变化的基因,比如
AKT_UP.V1_DN对应RAD001试剂处理后表达量下调的基因。 -
C7 : immunologic signatures
该类别包含了免疫系统功能相关的基因集合。
-