
简介
从细胞提取到的RNA序列中,其中占大部分(80%以上)的都是rRNA,这就是所说的“量大”。在转录组测序中,我们一般关注的是信使RNA(mRNA),因此,rRNA并不是目标序列,不去除rRNA的话,测序时会产生很多无用的rRNA序列数据,这就是所说的“不管饱”。
而且,就算是去掉了rRNA之后,mRNA的含量也不算是大头,因为还有其他的非编码RNA存在,比如tRNA、ncRNA等,但比起去除rRNA前已经好很多了。一般来说,转录组在实验阶段就会针对rRNA量大的问题进行处理,方法就我所知有两种类型:1、富集法(把目标搞出来),2、酶消化法/探针法(把非目标干掉)。
经过实验处理后,会大大降低产出中rRNA的比例,良好的产出中rRNA的占比可以降到1%以下,当然,拿到测序数据的时候,我们并不清楚,其中的rRNA的情况是怎么样的。在转录组测序中,个人认为低于10%的rRNA比例是可以接受的,只需要将其中的rRNA序列去除,将剩余的数据拿来做后续分析即可。而更高rRNA比例的转录组样本,说白了,其中的mRNA序列是不是随机得到的,都不清楚,虽然说后续分析可以质控,但良好的实验是结果的保证。
bowtie2去除rRNA
准备
- 转录组数据
- rRNA参考序列
- seqkit软件
- bowtie2软件
rRNA参考序列获取
-
NCBI的网站获取需要rRNA参考序列,输入
homo sapiens检索人类参考基因组信息,结果中有一个 “GCF_000001405.39”,即物种参考序列的ID号/版本号。 -
返回到NCBI的首页网站,下拉网页到最底部,进入NCBI FTP Site这个链接。
-
在站点里依次进入”genomes”-“all”-“GCF”-“000”-“001”-“405” 即刚才的参考序列版本号,进入人类的参考基因组。
-
GRCh37和GRCh38是常用的两个人类参考序列版本,也即对应着UCSC的hg19和hg38,我这边要用到的是GRCh37,选择了最新的p13版本,“GCF_000001405.25_GRCh37.p13”。
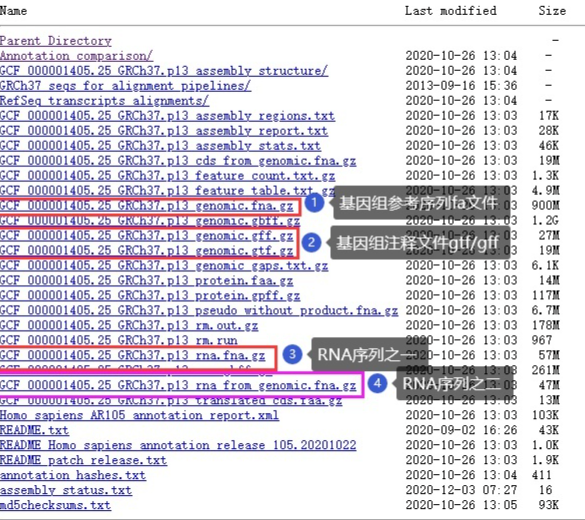
其中,①是参考基因组序列fa文件,②是基因组注释文件,③和④都是rna的序列,③和④的区别,我目前发现,④里有线粒体产生的RNA的序列,而③里没有,其他的不同,我没有去看。而我们需要的人类rRNA的序列就在④里,因为人类的12S、16SrRNA都是线粒体上的基因转录产生的,如果用③的话,rRNA序列将没有12S、16S这两条。将①~④都下载下来,应该还蛮快的,有时候网络或许很慢,重新下载就好了。
下载好人类rna序列之后解压,那么我们需要提取其中的rRNA序列,用作后续去除rRNA。
先查看下“GCF_000001405.25_GRCh37.p13_rna_from_genomic.fna”的序列id信息行,命令:
cat GCF_000001405.25_GRCh37.p13_rna_from_genomic.fna | grep "^>" | less -S可以看到,“gbkey”这个关键字提示这个rna是哪种rna,因此,可以考虑获取rRNA序列的id信息行,查看命令:
cat GCF_000001405.25_GRCh37.p13_rna_from_genomic.fna | grep "^>" | grep "gbkey=rRNA" | less -S -
得到rRNA的id信息行之后,你可以自己写脚本将rRNA序列提出来,也可以使用seqkit软件提取
-
提取rRNA的id,即id信息行的第一个空格前的字符串,输出到文本。(id信息行和id是不同的。)
-
linux下,解压后的seqkit是可以直接使用的,提取对应ID的序列,命令:
seqkit grep -f id.list GCF_000001405.25_GRCh37.p13_rna_from_genomic.fna > rRNA.fa
在NCBI上下载rRNA的fasta序列
-
打开NCBI,选择
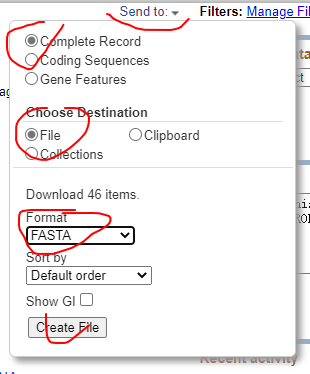
Taxonomy搜索homo-sapiens,点击Homo sapiens进入页面后,点击后边Nucleotide的Subtree links,再勾选左边rRNA,然后使用send to保存核糖体rRNA的序列。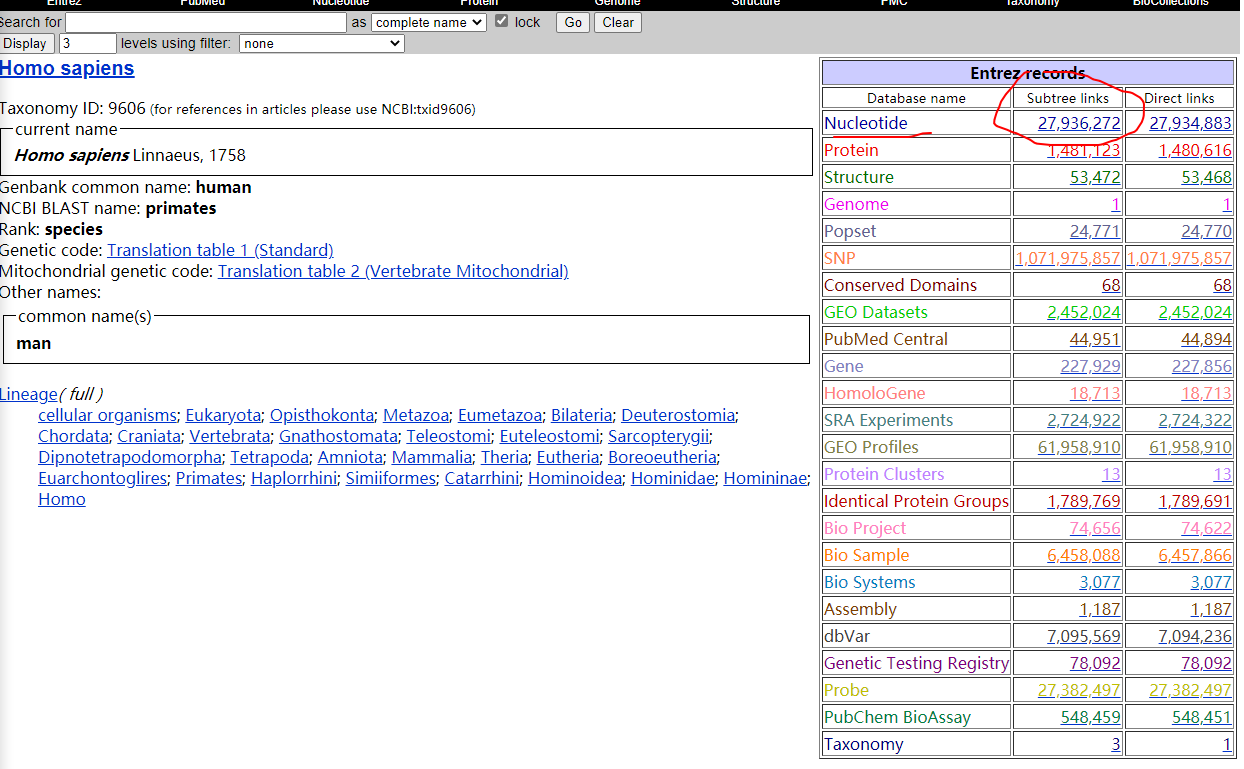
比对-bowtie2
-
安装bowtie2
conda install -y bowtie2 -
构建rRNA.fa的index
bowtie2-build rRNA.fa homo_sapiens_rRNA -
bowtie2去除rRNA
bowtie2 -x /public/jychu/reference/index/bowtie2/sheep/rRNA/sheep_rRNA -1 T_F2a_1_1.fq.gz -2 T_F2a_1_2.fq.gz --un-conc-gz T_F2a_input_rmrRNA.fastq.gz -p 8 -S T_F2a_rRNA.sam; rm T_F2a_rRNA.sam;bowtie2 --very-sensitive-local --no-unal -I 1 -X 1000 -p 6 -x rRNA -1 R1.fq.gz -2 R2.fq.gz --un-conc-gz sample_rRNAremoved.fq.gz 2>sample_Map2rRNAStat.xls | samtools view -S -b -o sample_Map2rRNA.bam -- –very-sensitive-local –no-unal -I 1 -X 1000:这几个参数不描述,可以看看bowtie2的帮助页;
- -p:线程数。
- -x:参考序列的前缀(就是bowtie2-build rRNA.fa rRNA中的第二个参数)。
- -1,-2:分别接测序数据的R1和R2,没有R2的话,只需要-1。
- –un-conc-gz:比对不上的序列,以此前缀输出。
- sample_Map2rRNAStat.xls是bowtie2最后给出的比对结果,包括比对率、唯一比对、多重比对率等。
-
后面可以直接通过管道命令“ ”,接samtools view -S -b -o sample_Map2rRNA.bam - 直接输出比对到rRNA的bam文件,用作其他分析。也可以不用samtools,直接 通过 “>”输出成 sam 文件。 - samtools的各个模块,常用的有view,sort,index,depth,mpileup,merge,基本上是生信用的最多的软件之一。
-
bowtie2跑完之后,将得到几个文件:

rRNAremoved的R1和R2的fq,这个就是我们最后想拿到的,去除了rRNA的序列,用于后续分析;Map2rRNA.bam,比对上rRNA的bam;sort和bai是我做了sort和index得到,不需要的可以不做;Map2rRNAStat.xls是比对到rRNA的情况。
示例
-
原始数据集fastqc质量问题:
-
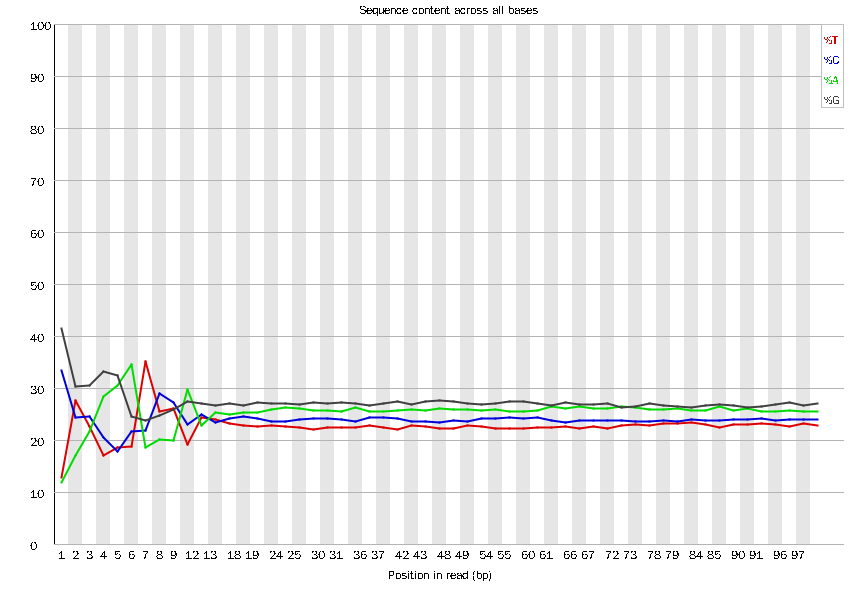
每个位置碱基质量较差
-
GC含量存在明显双峰,在80左右位置存在双峰
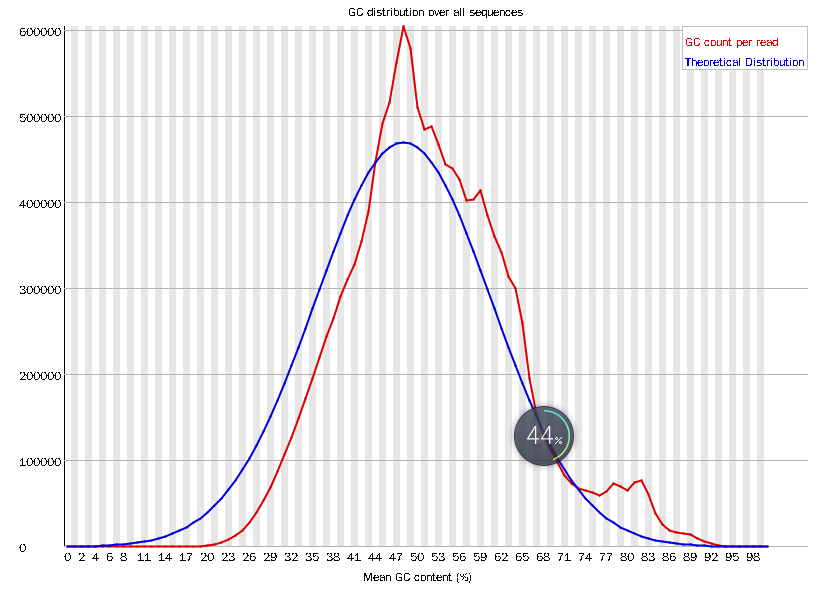
-
Sequence Duplication Levels
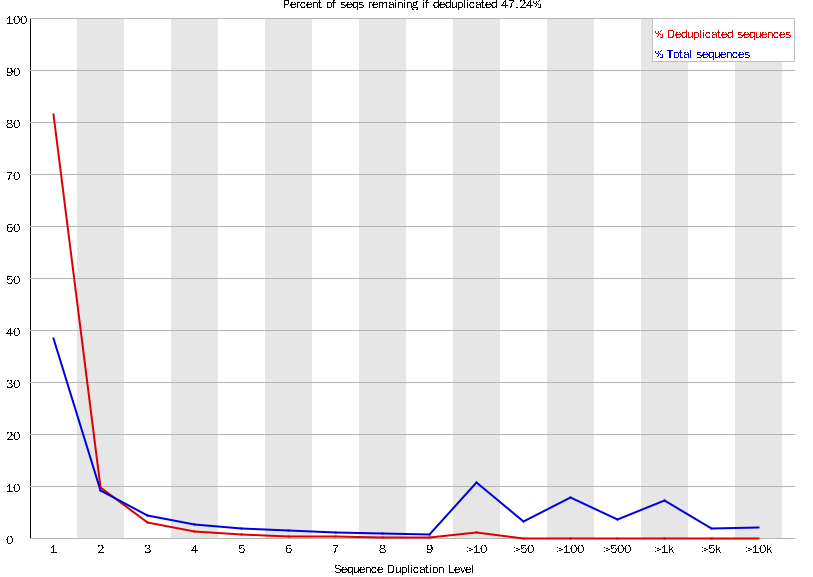
-
-
质控后的cleanData仍然没有改善
-
挑取GC含量双峰位置的部分reads blast,检查是什么污染

发现这些reads都可能与核糖体rRNA有关,因为怀疑存在核糖体rRNA污染
-
去除fastq数据中rRNA序列后fastqc
原始fastqc 序列数:14735820
去除rRNA 序列数: 11372630
非rRNA序列占比:77.2%
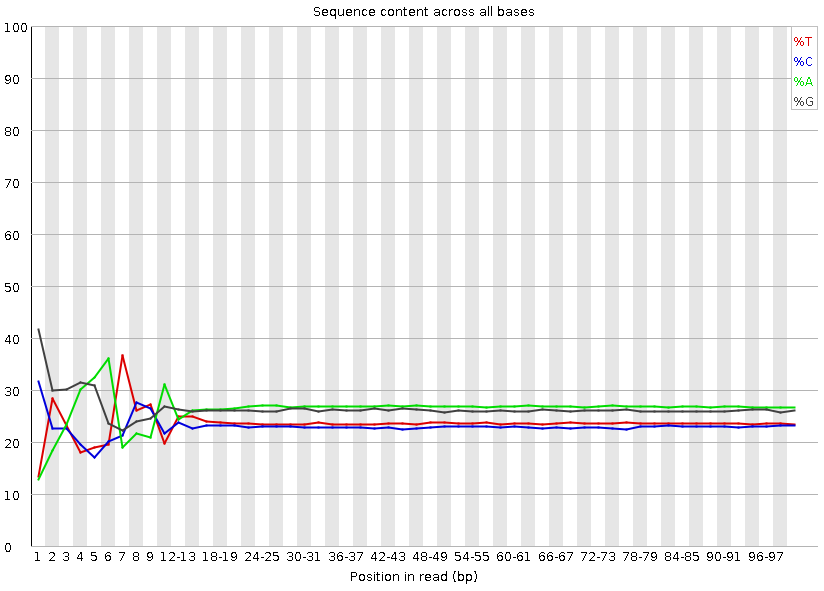
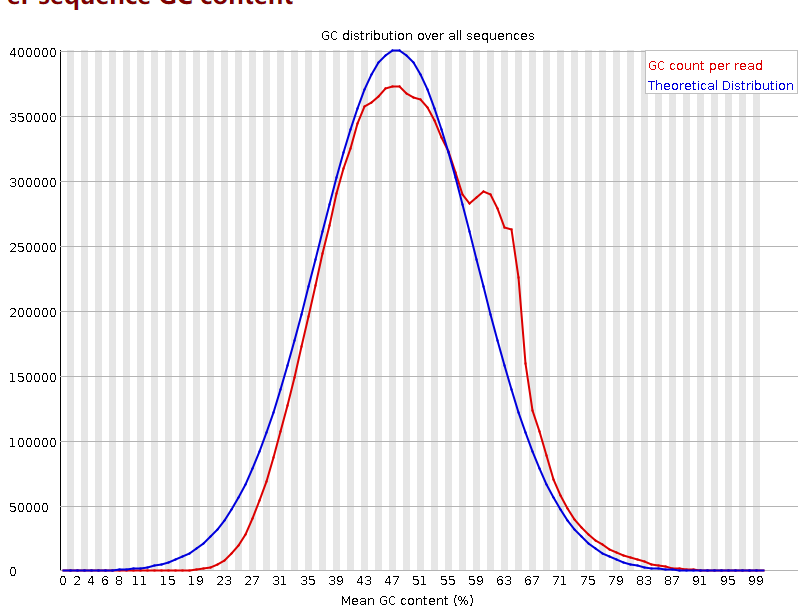
通过比较fastqc可以发现,去除rRNA后数据的碱基比例基本稳定,GC双峰基本移除。